

What Effects Do You Think Skeletal Disorders Have On Animals?
Introduction
Rare human genetic diseases cumulatively impact about 1 in 200 individuals and involve an estimated 7,000 genes. Major research efforts are underway to identify these mutant genes and characterize their disease phenotypes. Noesis gained tin can guide therapies and provide hypotheses to develop future treatments. As recently summarized (ane), "Genome sequencing has revolutionized the diagnosis of genetic diseases. Close collaborations between basic scientists and clinical genomicists are now needed to link genetic variants with disease causation. To facilitate such collaborations, we recommend prioritizing clinically relevant genes for functional studies, developing reference variant-phenotype databases, adopting phenotype description standards, and promoting data sharing."
Rare human genetic skeletal dysplasias affect about ane in 5,000 individuals (2) and account for 5% of all birth defects (iii). The International Skeletal Dysplasia Society (ISDS, https://www.isds.ch), promotes scientific progress in the field of skeletal dysplasias and dysostoses, meets every 2d year, and published skeletal nosology summaries during 2001 (four), 2006 (5), 2010 (6), 2015 (seven), and 2019 (eight). In that location are soon 441 skeletal nosology genes, with an boilerplate of 20 new genes identified yearly (Figure 1). The classification aims to (i) place metabolic pathways active in cartilage and os, and their regulatory mechanisms; (ii) place cellular signaling networks and gene expression sequences implicated in skeletal evolution; (iii) identify candidate genes for genetic disorders; (four) facilitate integration of data coming from spontaneous and genetically engineered mouse mutants; (v) assistance in developing diagnostic strategies; (vi) stimulate the design and exploration of new therapeutic possibilities; and (vii) provide a knowledge framework accessible to physicians likewise every bit to bones scientists and thus to facilitate communication between clinical genetics and pediatrics and the basic sciences (4).
Figure i
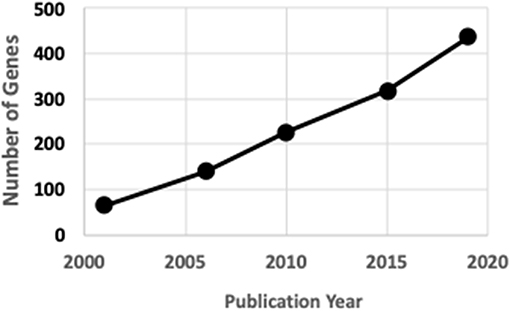
Figure 1. ISDS Nosology factor identification.
The objectives of the present review include further characterizations of these 441 skeletal nosology genes and evaluating the reliability of mutant mouse models to mimic these human skeletal disorders.
Historical Highlights
Brusque stature and other visually obvious skeletal dysplasias were apparent throughout human being history (9). The discovery of X-rays by Wilhelm Röntgen (10) was apace followed by the description of osteopetrosis past Albers-Schönberg (xi) and many skeletal dysplasias during the following decades (12). Dual-energy X-ray absorptiometry (DXA) engineering, developed during the 1980s (13), permitting quantitation of bone mineral density (BMD), and connected advances in computed tomography (CT), providing 3 dimensional images, lead to increasing sophisticated agreement of bone dysmorphology. The outset nosology gene identified was CA2 (carbonic anhydrase 2, osteopetrosis), initially in 1983 using electrophoretic, enzymatic and immunologic techniques on red blood cell extracts (xiv), and later by genetic mutation analysis in 1991 (fifteen). The starting time genetic mutation for whatever human disease to be identified past WES was DHODH (dihydroorotate dehydrogenase), responsible for postaxial acrofacial dysostosis, in 2010 (16).
Nosology
Nosology is the nomenclature of diseases, which in its simplest form involves symptoms and pathogenic mechanisms. No classification system is perfect and there are often multiple ways to classify a given disorder. At the extremes, "lumpers" and "splitters" adopt few and many categories, respectively (17). Heredity tin can exist X-linked, autosomal dominant, or autosomal recessive. Skeletal dysplasias tin can affect the skeleton only, or be part of pleiotropic syndromes affecting multiple organs. Mutations of various genes within a molecular pathway can each produce similar phenotypes. Loss-of office (LoF) mutations completely disrupt the activities of their encoded proteins just hypomorphic mutations allowing reduced protein activities occur. Gain-of-function (GoF) mutations increase the activities of enzymes and receptors and produce different phenotypes than LoF mutations. Dominant-negative mutations adversely touch functions of wild-type proteins. Mutations tin occur within the protein-coding region of the genome (exome), within introns, or betwixt gene coding regions. Mutations include deletions, duplications, and inversions.
The 2019 edition of the ISDS Nosology and Classification of Skeletal Disorders database organizes mutant human skeletal phenotypes into 42 groups, based on clinical observations and known gene/phenotype relationships (8). A total of 461 disorders and 441 genes are provided, when all x genes listed within the Notes sections of the tables (Tabular array 1) are included. Updated HGNU factor symbols for 11 genes (Table ii) are employed. Supplemental Table i provides an alphabetical listing in spreadsheet format of all 441 genes, with information on heredity, gene function and mouse model status. Genetic disorders are not listed, as mutations in many genes result in multiple phenotypes. Inheritance patterns are 242 autosomal recessive, 135 autosomal dominant, 34 autosomal recessive or autosomal dominant depending upon the exact mutation in the gene, 21 X-linked and 11 not-inherited, somatic mutations. 3 genes can take either germline or somatic mutations.
Table 1
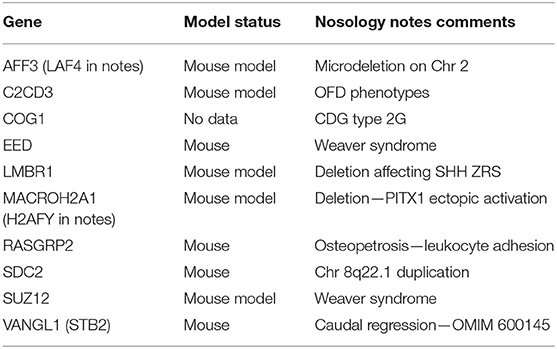
Table 1. Genes identified in 2019 Nosology notes section.
Tabular array 2
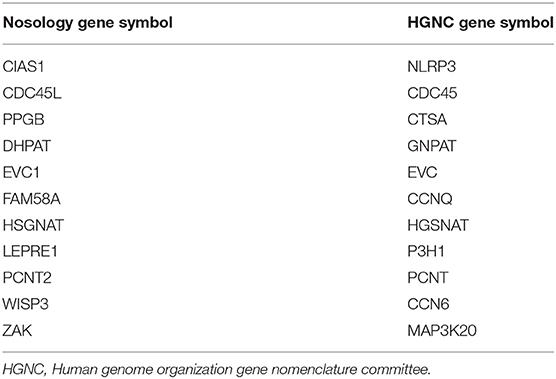
Table ii. Cistron symbol classification.
RMRP encodes an RNA regulating Dna transcription, RNU4ATAC encodes an RNA that is a component of an enzyme complex, and MIR140 is a microRNA. Proteins (and the 3 RNAs) function as enzymes (146, 33%), scaffold components (79, xviii%), ligand/receptor signaling molecules (72, 16%), transcription factors (62, 14%), cilia components (36, 8%), matrix proteins (23, 5%), membrane transporters (19, 4%), and cohesionopathy proteins (4, 1%). These eight gene role categories are informative simply arbitrary, and other categories tin can be envisioned. For example, 23 enzymes are involved in the synthesis, processing, and deposition of protein and glycosaminoglycan matrix components. Skeletal disorders include malfunctions of lysosomal function. Signaling genes can be assigned to BMP, FGF, WNT, and other pathways.
In that location are no orthologous mouse genes for man ARSE (arylsulfatase E) and RNU4ATAC (RNA, U4atac small nuclear, U12-dependent splicing). Supplemental Table one summarizes published information on the availability and fidelity of mouse models for the 439 human rare bone affliction genes. Mutant mice with bone phenotypic data exist for 260 of the 439 genes (59%) with similar bone phenotypes observed for 249 (96%) genes. Supplemental Table 2 contains PubMed hyperlinks to publications for all 249 genes provided in Supplement Table 1 having mutant mouse bone phenotypes. These two supplemental tables should provide a major resource for the bone research community.
Mutant mouse bone information are inconsistent with human skeletal phenotypes for 11 genes (Ccn6, Cyp2r1, Flna, Galns, Gna13, Lemd3, Manba, Mnx1, Nsd1, Plod1, Smarcal1). There are no obvious explanations for or commonalities among these human-mouse phenotype inconsistencies. For 97 genes (22%) mutant mice have been generated and examined, just no skeletal information were reported. Mutant mice do not appear to have been examined for 82 genes (19%) and 36 (eight%) of these genes belong to the understudied Ignorome/Dark Genome (xviii–xx). Individual laboratories and/or consortia are encouraged to examine these genes, at present known to contribute to poorly understood human rare os diseases.
The number of os nosology genes continues to increase as novel genes affecting skeletal metabolism are identified in homo subjects. The genes described in this report form an arbitrary "snapshot" taken during August 2019 and will undoubtedly increase. Skeletal disorders for which mutant genes have not been identified include CDAGS syndrome (OMIM 603116), cherubism with gingival fibromatosis (OMIM 266270), chondrodysplasia punctata tibial-metacarpal blazon (OMIM 118651), dysplasia epiphysealis hemimelica (OMIM 127800), femur fibula ulna syndrome (OMIM 228200), hemifacial microsomia (OMIM 1642100, genochondromtosis (OMIM 1373600, Moreno–Nishimura–Schmidt syndrome (OMIM 608811), pachydermoperiostosis (OMIM 167100), and thoracolaryngopelvic dysplasia (OMIM 187760).
Germination of a normal skeleton involves BMP, FGF, and WNT signaling pathways and mutations in multiple genes within these pathways ofttimes produce skeletal dysplasias. Bone cells respond to parathyroid hormone, the active vitamin D metabolite calcitriol, and circulating FGF23 as part of the calcium-phosphate homeostatic system and disruptions in these hormones produce skeletal endocrinopathies. Skeletal disorders involving aggrecanopathies (13), channelopathies (21), ciliopathies (22, 23), cohesinopathies (24), lamiopathies (25), linkeropathies (26), protein-folding defects (27), ribosomopathies (28), spliceosomopathies (29), and transcription factors (30) show the importance of pathways not often thought to be involved in bone development.
Skeletal Disorder Vignettes
This department briefly summarizes selected skeletal disorders resulting from various mutations, highlighting the broad range of transcription and translation events that tin can exist disrupted.
• Mutations can be beneficial with healthy nutrition only produce affliction when cardinal nutrients are lacking. All humans have an inactivating mutation in GULO, encoding an enzyme involved in the synthesis of ascorbic acid, and develop scurvy without sufficient dietary intake of vitamin C. The ascorbate synthetic pathway, involving aldehyde and aldose reductases, was only fully characterized in 2010 (31). Ascorbic acid is a required cofactor for the hydroxylation of proline and lysine residues in collagen and disruption of the mouse gulonolactone oxidase gene results in spontaneous bone fractures (32). Similarly, human and mouse HAAO and KYNU genes are involved in the synthesis of the enzymatic cofactor NAD and inactivating mutations in these human and mouse genes can result in congenital malformations (33).
• X-linked human mutations comprise half-dozen% of the total skeletal disorders. X-inactivation of one of the ii 10 chromosomes in women by long non-coding RNA specific transcript XIST occurs, but nearly 20% of X chromosome genes escape this inactivation (34). AMER1 and PORCN are Ten-linked genes that code for components of the WNT signaling pathway, with dominant mutations in women causing osteopathia striata with cranial sclerosis and focal dermal hypoplasia (including osteopathia striata), respectively. Due to developmental lethality male patients are extremely rare, merely a few males having post-zygotic mosaic mutations have been identified (35, 36). Amer1 mutations in mice disrupt bone architecture (37) and treating adult mice with inhibitors of the PORCN enzyme reduces bone mass (38).
• Somatic gene mutations in 11 genes (AKT1, FLBN, GNAS, GREM1, HRAS, IDH1, IDH2, MAP2K1, NOTCH2, NRAS, PIK3CA) arise in the developing zygote and are non transmitted genetically. Loeys-Dietz syndrome includes several skeletal dysplasias and can result from mutations in SMAD2, SMAD3, TGFB2, TGFB3, TGFBR1, or TGFBR2 and 75% of afflicted subjects have somatic mutations (39). Melorheostotic, dense hyperostotic bone lesions are caused by somatic mosaic mutations in KRAS (40) and MAP2K1 (41). MAP2K1 mutations are idea to arise after the formation of dorso-ventral plane (42). KRAS and MAP2K1 are non included amidst the 441 Nosology disorders. Mutations in COL11Ai, EZH2, and MET can be either germline or somatic.
• Deleterious mutations can occur at multiple sites within genes. For example, in that location are 1053 COL1A1 DNA variants in the Osteogenesis Imperfecta Variant Database as of September 2019 (https://oi.gene.le.air conditioning.uk/home.php?select_db=COL1A1, accessed 13 December, 2019).
• Splicing mutations that disrupt normal exon transcription within the spliceosome are estimated to contribute to 15% of man genetic diseases (43, 44). Acrofacial and mandibulofacial dysostosis often involve spliceosome defects and mutations in EFTUD2, EIF4A3, and SF3B4 genes each result in distinct craniofacial phenotypes. Splice site mutations in AIFM1 (45), SERPINF1 (46), and TRAPPC2 (47) result in skeletal dysplasias.
• MicroRNAs are non-protein coding unmarried-stranded RNAs (48) that regulate gene expression in os and other tissues. Mouse studies show that microRNA-140 is involved in growth plate development (49, l). A gain-of function mutation in microRNA-140 results in human being skeletal dysplasia (51).
• Subjects with intragenic duplications of IFT81 (tandem duplication of exons 9 and 10) and MATN3 (tandem duplication of exons two–five), detected by WGS, have skeletal dysplasias similar to subjects with LoF mutations in these genes (52).
• Autosomal-dominant syndactyly, synpolydactyly, and brachydactyly types D and Due east can outcome from ascendant-negative mutations in the homeobox cistron HOXD13. Duplications of the HOXD gene cluster locus produce mesomelic dysplasia with shortened limbs (53, 54). Similar Hoxd locus GoF alterations in ulnaless mutant mice, generated by Ten-irradiation, produce similar bone phenotypes (55, 56).
• ISDS nosology includes skeletal disorders resulting from disruptions of calcium-phosphate homeostasis, including various endocrinopathies. Regulation of calcium and phosphorus homeostasis involves ALPL, CASR, DMP1, ENPP1, FAM20C, FGF23, GALANT3, HRAS, KL, NRAS and TRPV6 genes. Parathyroid hormone synthesis and action involve CDC73, FAM111A, GCM and PTH1R. Vitamin D synthesis and deportment involve CYP2R1, CYP27B1 and VDR. Normal Ca and P homeostasis occurs in humans (57) and mice (58) with deletions of the GC cistron and thereby defective the circulating vitamin D-binding protein (DBP) that binds serum 25-OH-D. Multiple neonatal bone fractures were observed due to maternal hypoparathyroidism and vitamin D deficiency (59).
Heredity of Bone Mass Without Skeletal Dysplasia
Osteoporosis is a common skeletal disease in which reduced amounts of otherwise normal bone pb to fragility and fractures. Developed bone mass, even within the normal range, has a potent heredity influence (threescore, 61) and identifying the genes involved in os mass accumulation during growth and loss during aging has received great interest within the context of the etiology and treatment of osteoporosis. GWAS studies over the past decade described an increasing number of genes affecting BMD, with 518 loci identified in the 2019 UK Biobank analysis (62). Juvenile osteoporosis, although not a true dysplasia equally bone architecture is normal, normally has genetic causes (63, 64). In that location are healthy subjects with unexplained loftier bone mass (65, 66) and attempts are underway to identify the genes responsible. Contempo discoveries of such genes include LRP6 (67) and SMAD9 (68).
Mouse Models
All models are wrong, just some are less imperfect than others, and many are useful - George Box
Mouse models make important contributions to understanding and treating human diseases (69–72), including skeletal disorders (73, 74). Mutant mice that model homo phenotypes as well model successful drugs (75), help identify genes responsible for homo genetic disorders and can provide insights for osteoporosis drug evolution (76). Bone mass and architecture vary in good for you humans and amid laboratory mouse strains, with the most commonly studied C57BL/6 mouse strain an outlier having limb bones with loftier diameters and depression cortical thickness (77–81).
The majority of mouse information summarized in this review involve individual investigator-initiated studies examining possible skeletal phenotypes in transgenic mice with specific alterations in genes chosen by the investigator. This approach, known as opposite genetics, utilizes the expertise of the laboratories involved.
In dissimilarity, human studies involve forrard genetics, with genes responsible for known skeletal phenotypes identified. Forrad genetics is as well employed in mouse studies, as genes responsible for spontaneous and mutagen-induced skeletal malformations are identified. The Jackson Laboratories (JAX), with a long history of studying mouse strains, recently employed WES to place 14 genes having spontaneous mutations causing bone phenotypes (82, 83). Several laboratories employed Due north-ethyl-N-nitrosourea (ENU) in chemical mutagenesis campaigns to produce mouse lines having a wide-range of phenotypes. This approach yielded 41 genes having mutations causing os phenotypes similar to the corresponding human skeletal disorders. These 41 genes with relevant citations are provided in Supplemental Table 3.
2 loftier-throughput mouse reverse genetics gene knockout phenotyping campaigns take been undertaken (84). The International Mouse Phenotyping Consortium (IMPC, world wide web.mousephenotype.org) aims to characterize knockout mouse phenotypes for all twenty,000 genes (74, 85). Lexicon Pharmaceuticals' Genome5000™ effort examining the druggable genome confirmed known bone phenotypes for 23 genes and identified eleven genes, including Notum (86), for which bone phenotypes were not previously characterized (87). Importantly, skeletal phenotypes were described for Fam20c (non-lethal Raine syndrome), Lrrk1 (osteosclerotic metaphyseal dysplasia), Pappa2 (brusk stature), Sfrp4 (Pyle'southward disease), and Slc10a7 (skeletal dysplasia) prior to cognition of the human skeletal dysplasias when mutated in humans (84). For the 439 mouse genes discussed in this review, 149 genes have been examined by the IMPC, yielding 63 feasible developed homozygous mouse mutants. Skeletal phenotypes (either torso BMD or radiological dysmorphology) were observed for 28 genes. Results from the IMPC phenotyping campaign are summarized in Table iii.
TABLE 3

Table 3. Summary of International Mouse Phenotyping Campaign (IMPC) models.
Mouse models of human genetic disorders are employed to evaluate potentially beneficial skeletal actions of therapies approved for other illness indications. Teriparatide treatment increases os mass in Lrp5 KO mice mimicking humans with osteoporosis pseudoglioma syndrome from loss of function LRP5 mutations (88, 89). Similarly, anti-sclerostin antibiotic treatment increases bone mass in mutant mouse models with low bone mass from gene disruptions (90) of Col1a1 (91, 92), Col1a2 (93, 94), Crtap (95), Dmp1 (96), Lrp5 (97), and Zmpste24 (98). Mechanistic hypotheses can be tested, such equally periostin handling retarding skull suture fusion in heterozygous Twist1 mice with craniosynostosis (99).
Mouse Study Precautions
Several experimental pitfalls should exist avoided when performing mouse studies (100).
• Knockout of private genes can disrupt the functions of neighboring genes (101). Examples include the presence of orofacial defects resulting from a hypomorphic Pax9 allele during knockout of the neighboring Slc25a21 cistron (102) and glycosaminoglycan accumulation resulting from reduced expression of the Naglu cistron during knockout of the neighboring Hsd17b1 gene (103).
• Transgenic Cre mouse lines are invaluable for conditionally activating or inactivating genes of interest. Several reporter genes are bachelor for visualizing os cells at different stages of evolution (104). But not all Cre lines are as specific as originally believed (105–107). Understanding these limitations is critical for experimental pattern and estimation.
• Quantitative PCR methods are oft not optimized and MIQE (Minimum Information for the publication of qPCR Experiments) guidelines have been established (108, 109). Selection of the advisable reference gene(southward) is important (110–112).
• Many antibodies suffer from a lack of specificity resulting from cross-reactivity to similar epitopes nowadays on multiple proteins. Clifford Saper in 2005, as Editor-in-Master of The Periodical of Comparative Neurology, repeatedly received "… distressed communications from authors … to withdraw papers considering an antibody against a novel marker was found to stain tissue in knockout animals …" (113). Splendid reviews (not cited here) provide guidelines for successful antibiotic validation and the purposeful joviality in their titles ("Antibody Tin Go It Right … Antibody Anarchy … Antibody Crimes … A Guide to the Perplexed … Garbage In, Garbage Out … Hitchhiker Antigens … Non for the Faint-Hearted … The Nighttime Side of the Immunohistochemical Moon … The Proficient, Bad, and Actually Ugly") emphasizes the seriousness of the problem. Antibodies claimed to be specific for item proteins should not react against tissues from KO mice missing the factor of interest and validation of antibody specificity using tissues from KO cells or mice is strongly encouraged.
• Established cell lines employed in conjunction with mouse studies tin can become contaminated and replaced past more than robust, faster growing cells (114). Cell line hallmark methods exist and should be employed (115, 116). MC3T3-E1 cell subclones vary as models of osteoblast biology (117).
Large Fauna and Zebrafish Models
Large animals can take advantages over rodents for agreement homo genetic disease and drug development. Hypophosphatasia occurs in sheep (118) and dogs (119) having mutations in ALPL. Canine genetic skeletal disorders include mutations in ADAMTSL2—geleophysic dysplasia (120), COL1A2—osteogenesis imperfecta (121), DVL2—Robinow syndrome (122), HES7—spondylocostal dysostosis (123), and SERPINH1—osteogenesis imperfecta (124). Spontaneous mutations in chicken KIAA0586 (125) and LMBR1 (126) genes result in the expected bone phenotypes.
Zebrafish are increasing contributing to our cognition of skeletal genomics (127, 128) and advantages over mouse models include acquiring information more chop-chop. Zebrafish mutants have been described for several of the 441 genes in this review. One complication of zebrafish studies is that zebrafish underwent a teleost-specific whole genome duplication and have more than than 26,000 protein-coding genes (129). There is a one-to-one relationship betwixt 47% of human genes and a zebrafish ortholog. In that location are multiple zebrafish genes associated to a single human gene, and vice versa.
Drug Evolution
Exciting advances are existence made in developing drug treatments for patients with genetic skeletal disorders (130, 131) and mouse models invariably contribute to this progress. These advances are best reviewed by the laboratories involved, but three examples are illustrative. An antibody to NOTCH2 reverses osteopenia in a mouse model of Hajdu-Cheney syndrome (132). Cinacalcet corrects hypercalcemia in a mouse model of familial hypercalcemia type 2 (133). ENPP1 enzyme replacement therapy improves claret pressure and cardiovascular function in a mouse model of generalized arterial calcification of infancy (134).
Agreement genetic skeletal disorders provides key cognition for developing osteoporosis therapies (76, 135). Disruptions in genes coding for proteins in the RANK—RANKL—osteoprotegerin signaling pathway involved in osteoclast generation cause human skeletal disorders. The RANKL neutralizing antibiotic denosumab is a successful osteoporosis therapy. The recently approved anabolic osteoporosis handling romosozumab, a sclerostin neutralizing antibody, was developed with knowledge gained from subjects with osteosclerosis resulting from SOST gene mutations. Subjects with pinocytosis have mutations in the cathepsin K coding gene CTSK. Treatment with odanacatib, an inhibitor of cathepsin K in osteoclasts, reduced bone fractures in postmenopausal women but cardiovascular side effects precluded regulatory blessing.
Futurity Directions
Since many human disorders involve hypomorphic, gain-of-role, dominant-negative and intronic mutations, future studies will undoubtedly employ CRISPR/Cas9 technology and other evolving techniques to examine transgenic mice having genes modified to exactly mimic variant man sequences (72, 136). RNA sequencing will increasingly be employed for diagnosis and mechanistic agreement of genetic diseases (137–141).
The IFMRS (International Federation of Musculoskeletal Research Societies), in collaboration with the Wide Institute, is establishing a Musculoskeletal Genomics Noesis Portal (MGKP) to integrate, interpret and nowadays human data linked to musculoskeletal traits and diseases (http://world wide web.kp4cd.org/nigh/os).
Writer Contributions
RB performed the literature search, data analyses, and prepared the manuscript. CO provided helpful suggestions and reviewed the manuscript.
Funding
This study was supported by funding from the Swedish Research Council (Grant 2016-01001), ALF/LUA research grant from the Sahlgrenska Academy Hospital, Lundberg Foundation, the Torsten Söderberg Foundation, The Knut and Alice Wallenberg'southward Foundation and the Novo Nordisk Foundation.
Disharmonize of Interest
The authors declare that the enquiry was conducted in the absence of any commercial or fiscal relationships that could be construed as a potential disharmonize of interest.
Acknowledgments
Matt Warman (Boston Children's Hospital) encouraged this project. Scott Youlten (Garvan Institute of Medical Research) independently extracted gene IDs from the 2019 ISDS Nosology paper and provided Ensembl gene IDs.
Supplementary Material
The Supplementary Material for this article tin be plant online at: https://www.frontiersin.org/manufactures/x.3389/fendo.2019.00934/total#supplementary-material
References
1. Manolio TA, Fowler DM, Starita LM, Haendel MA, MacArthur DG, Biesecker LG, et al. Bedside dorsum to bench: building bridges between basic and clinical genomic research. Cell. (2017) 169:vi–12. doi: 10.1016/j.cell.2017.03.005
PubMed Abstract | CrossRef Total Text | Google Scholar
6. Warman ML, Cormier-Daire V, Hall C, Cracow D, Lachman R, LeMerrer Thou, et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A. (2011) 155A:943–68. doi: x.1002/ajmg.a.33909
PubMed Abstract | CrossRef Full Text | Google Scholar
vii. Bonafé Fifty, Cormier-Daire V, Hall C, Lachman R, Mortier Grand, Mundlos S, et al. Nosology and nomenclature of genetic skeletal disorders: 2015 revision. Am J Med Genet A. (2015) 167A:2869–92. doi: 10.1002/ajmg.a.37365
PubMed Abstruse | CrossRef Full Text | Google Scholar
8. Mortier GR, Cohn DH, Cormier-Daire V, Hall C, Krakow D, Mundlos Due south, et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A. (2019) 179:2393–419. doi: 10.1002/ajmg.a.61366
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Albers-Schönberg HE. Röntgenbilder einer seltenen Knochenerkrankung. Münchener Medizinische Wochenschrift. (1904) 51:365–76.
Google Scholar
xiii. Lewiecki EM, Binkley N. DXA: 30 years and counting: Introduction to the 30th anniversary event. Bone. (2017) 104:1–3. doi: ten.1016/j.bone.2016.12.013
CrossRef Full Text | Google Scholar
xiv. Sly WS, Hewett-Emmett D, Whyte MP, Yu YS, Tashian RE. Carbonic anhydrase Two deficiency identified as the primary defect in the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. Proc Natl Acad Sci USA. (1983) 80:2752–6. doi: 10.1073/pnas.eighty.9.2752
PubMed Abstruse | CrossRef Full Text | Google Scholar
15. Venta PJ, Welty RJ, Johnson TM, Sly WS, Tashian RE. Carbonic anhydrase II deficiency syndrome in a Belgian family is caused past a indicate mutation at an invariant histidine residue (107 His-Tyr): complete structure of the normal homo CA II gene. Am J Hum Genet. (1991) 49:1082–90.
PubMed Abstract | Google Scholar
xvi. Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Paring KM, Huff CD, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. (2010) 42:xxx–five. doi: ten.1038/ng.499
PubMed Abstract | CrossRef Full Text | Google Scholar
17. McKusick VA. On lumpers and splitters, or the nosology of genetic affliction. Perspect Biol Med. (1969) 12:298–312. doi: 10.1353/pbm.1969.0039
CrossRef Full Text | Google Scholar
19. Oprea TI, Bologa CG, Brunak S, Campbell A, Gan GN, Gaulton A, et al. Unexplored therapeutic opportunities in the human genome. Nat Rev Drug Discov. (2018) 17:317–32. doi: ten.1038/nrd.2018.fourteen
CrossRef Full Text | Google Scholar
20. Stoeger T, Gerlach M, Morimoto RI, Nunes Amaral LA. Large-calibration investigation of the reasons why potentially important genes are ignored. PLoS Biol. (2018) 16:e2006643. doi: x.1371/periodical.pbio.2006643
PubMed Abstruse | CrossRef Full Text | Google Scholar
21. Gibson BG, Briggs Doc. The aggrecanopathies; an evolving phenotypic spectrum of human genetic skeletal diseases. Orphanet J Rare Dis. (2016) 11:86. doi: 10.1186/s13023-016-0459-two
PubMed Abstruse | CrossRef Total Text | Google Scholar
23. Yuan X, Serra RA, Yang S. Function and regulation of primary cilia and intraflagellar transport proteins in the skeleton. Ann NY Acad Sci. (2015) 1335:78–99. doi: 10.1111/nyas.12463
PubMed Abstract | CrossRef Total Text | Google Scholar
24. Cohen-Zinder Thou, Karasik D, Onn I. Structural maintenance of chromosome complexes and bone development: the beginning of a wonderful relationship? Bonekey Rep. (2013) 2:388. doi: 10.1038/bonekey.2013.122
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Ritelli M, Cinquina V, Giacopuzzi E, Venturini One thousand, Chiarelli Due north, Colombi M. Further defining the phenotypic spectrum of B3GAT3 mutations and literature review on linkeropathy syndromes. Genes. (2019) ten:E631. doi: 10.3390/genes10090631
PubMed Abstruse | CrossRef Full Text | Google Scholar
28. Trainor PA, Merrill AE. Ribosome biogenesis in skeletal evolution and the pathogenesis of skeletal disorders. Biochim Biophys Acta. (2014) 1842:769–78. doi: x.1016/j.bbadis.2013.11.010
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Lehalle D, Wieczorek D, Zechi-Ceide RM, Passos-Bueno MR, Lyonnet S, Amiel J, Gordon CT. A review of craniofacial disorders acquired by spliceosomal defects. Clin Genet. (2015) 88:405–15. doi: ten.1111/cge.12596
PubMed Abstract | CrossRef Full Text | Google Scholar
thirty. Chatterjee South, Sivakamasundari V, Lee WJ, Chan HY, Lufkin T. Making no bones about information technology: transcription factors in vertebrate skeletogenesis and disease. Trends Dev Biol. (2012) 6:45–52.
PubMed Abstract | Google Scholar
31. Gabbay KH, Bohren KM, Morello R, Bertin T, Liu J, Vogel P. Ascorbate synthesis pathway: dual role of ascorbate in bone homeostasis. J Biol Chem. (2010) 285:19510–xx. doi: 10.1074/jbc.M110.110247
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Aghajanian P, Hall South, Wongworawat Doctor, Mohan S. The roles and mechanisms of deportment of vitamin C in bone: new developments. J Bone Miner Res. (2015) 30:1945–55. doi: x.1002/jbmr.2709
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Shi H, Enriquez A, Rapadas M, Martin EMMA, Wang R, Moreau J, et al. NAD deficiency, congenital malformations, and niacin upplementation. N Engl J Med. (2017) 377:544–52. doi: 10.1056/NEJMoa1616361
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Tukiainen T, Villani AC, Yen A, Rivas MA, Marshall JL, Satija R, et al. Landscape of X chromosome inactivation across human tissues. Nature. (2017) 550:244–viii. doi: 10.1038/nature24265
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Vreeburg 1000, van Geel M, van den Heuij LG, Steijlen PM, van Steensel MA. Focal dermal hypoplasia in a male person patient due to mosaicism for a novel PORCN single nucleotide deletion. J Eur Acad Dermatol Venereol. (2011) 25:592–5. doi: x.1111/j.1468-3083.2010.03782.x
PubMed Abstract | CrossRef Full Text | Google Scholar
37. Comai G, Boutet A, Tanneberger G, Massa F, Rocha AS, Charlet A, Panzolini C, et al. Genetic and molecular insights into genotype-phenotype relationships in Osteopathia Striata with Cranial Sclerosis (OSCS) through the analysis of novel mouse Wtx mutant alleles. J Bone Miner Res. (2018) 33:875–87. doi: 10.1002/jbmr.3387
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Funck-Brentano T, Nilsson KH, Brommage R, Henning P, Lerner UH, Koskela A, et al. Porcupine inhibitors impair trabecular and cortical os mass and strength in mice. J Endocrinol. (2018) 238:13–23. doi: 10.1530/JOE-18-0153
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Loeys BL, Dietz HC. Loeys-Dietz Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens Grand, Amemiya A, editors. GeneReviews® . Seattle, WA: University of Washington (1993–2019).
Google Scholar
xl. Whyte MP, Griffith Thousand, Trani 50, Mumm S, Gottesman GS, McAlister WH, et al. Melorheostosis: exome sequencing of an associated dermatosis implicates postzygotic mosaicism of mutated KRAS. Bone. (2017) 101:145–55. doi: 10.1016/j.bone.2017.04.010
PubMed Abstract | CrossRef Full Text | Google Scholar
41. Fratzl-Zelman N, Roschger P, Kang H, Jha Southward, Roschger A, Blouin S, et al. Melorheostotic bone lesions acquired past somatic mutations in MAP2K1 have deteriorated microarchitecture and periosteal reaction. J Bone Miner Res. (2019) 34:883–95. doi: x.1002/jbmr.3656
PubMed Abstract | CrossRef Total Text | Google Scholar
42. Jha Due south, Laucis N, Kim 50, Malayeri A, Dasgupta A, Papadakis GZ, et al. CT analysis of anatomical distribution of melorheostosis challenges the sclerotome hypothesis. Bone. (2018) 117:31–6. doi: 10.1016/j.bone.2018.09.005
PubMed Abstract | CrossRef Total Text | Google Scholar
43. Park Eastward, Pan Z, Zhang Z, Lin L, Xing Y. The expanding mural of alternative splicing variation in human populations. Am J Hum Genet. (2018) 102:11–26. doi: x.1016/j.ajhg.2017.11.002
PubMed Abstruse | CrossRef Full Text | Google Scholar
45. Miyake Northward, Wolf NI, Cayami FK, Crawford J, Bley A, Bulas D, et al. Neurogenetics X-linked hypomyelination with spondylometaphyseal dysplasia (H-SMD) associated with mutations in AIFM1. Neurogenetics. (2017) xviii:185–94. doi: 10.1007/s10048-017-0520-x
CrossRef Full Text | Google Scholar
46. Jin Z, Burrage LC, Jiang MM, Lee YC, Bertin T, Chen Y, et al. Whole-exome sequencing identifies an intronic cryptic splice site in SERPINF1 causing osteogenesis imperfecta Type Half-dozen. JBMR Plus. (2018) 2:235–9. doi: 10.1002/jbm4.10044
PubMed Abstract | CrossRef Total Text | Google Scholar
47. Fukuma M, Takagi M, Shimazu T, Imamura H, Yagi H, Nishimura G, et al. A familial instance of spondyloepiphyseal dysplasia tarda caused by a novel splice site mutation in TRAPPC2. Clin Pediatr Endocrinol. (2018) 27:193–half dozen. doi: 10.1297/cpe.27.193
PubMed Abstruse | CrossRef Total Text | Google Scholar
49. Nakamura Y, Inloes JB, Katagiri T, Kobayashi T. Chondrocyte-specific microRNA-140 regulates endochondral bone development and targets Dnpep to attune bone morphogenetic poly peptide signaling. Mol Cell Biol. (2011) 31:3019–28. doi: 10.1128/MCB.05178-11
PubMed Abstract | CrossRef Full Text | Google Scholar
fifty. Papaioannou G, Mirzamohammadi F, Lisse TS, Nishimori S, Wein MN, Kobayashi T. MicroRNA-140 provides robustness to the regulation of hypertrophic chondrocyte differentiation by the PTHrP-HDAC4 pathway. J Bone Miner Res. (2015) thirty:1044–52. doi: 10.1002/jbmr.2438
PubMed Abstract | CrossRef Full Text | Google Scholar
51. Grigelioniene G, Suzuki How-do-you-do, Taylan F, Mirzamohammadi F, Borochowitz ZU, Ayturk UM, et al. Gain-of-function mutation of microRNA-140 in human skeletal dysplasia. Nat Med. (2019) 25:583–90. doi: ten.1038/s41591-019-0353-2
PubMed Abstruse | CrossRef Full Text | Google Scholar
52. Pettersson M, Vaz R, Hammarsjö A, Eisfeldt J, Carvalho CMB, Hofmeister W, et al. Alu-Alu mediated intragenic duplications in IFT81 and MATN3 are associated with skeletal dysplasias. Hum Mutat. (2018) 39:1456–67. doi: ten.1002/humu.23605
PubMed Abstract | CrossRef Full Text | Google Scholar
53. Kantaputra PN, Klopocki E, Hennig BP, Praphanphoj V, Le Caignec C, Isidor B, et al. Mesomelic dysplasia Kantaputra type is associated with duplications of the HOXD locus on chromosome 2q. Eur J Hum Genet. (2010) 18:1310–xiv. doi: 10.1038/ejhg.2010.116
PubMed Abstruse | CrossRef Total Text | Google Scholar
54. Le Caignec C, Pichon O, Briand A, de Courtivron B, Bonnard C, Lindenbaum P, et al. Fryns type mesomelic dysplasia of the upper limbs acquired by inverted duplications of the HOXD factor cluster. Eur J Hum Genet. (2019). doi: x.1038/s41431-019-0522-2. [Epub alee of impress].
PubMed Abstract | CrossRef Full Text | Google Scholar
55. Peichel CL, Prabhakaran B, Vogt TF. The mouse Ulnaless mutation deregulates posterior HoxD factor expression and alters appendicular patterning. Development. (1997) 124:3481–92.
PubMed Abstract | Google Scholar
56. Hérault Y, Fraudeau N, Zákány J, Duboule D. Ulnaless (Ul), a regulatory mutation inducing both loss-of-function and gain-of-function of posterior Hoxd genes. Development. (1997) 124:3493–500.
PubMed Abstract | Google Scholar
57. Henderson CM, Fink SL, Bassyouni H, Argiropoulos B, Chocolate-brown L, Laha TJ, et al. Vitamin D-binding poly peptide deficiency and homozygous deletion of the GC gene. Due north Engl J Med. (2019) 380:1150–seven. doi: ten.1056/NEJMoa1807841
PubMed Abstract | CrossRef Full Text | Google Scholar
58. Safadi FF, Thornton P, Magiera H, Hollis BW, Gentile M, Haddad JG, et al. Osteopathy and resistance to vitamin D toxicity in mice null for vitamin D binding protein. J Clin Invest. (1999) 103:239–51. doi: 10.1172/JCI5244
PubMed Abstract | CrossRef Full Text | Google Scholar
59. Alikasifoglu A, Gonc EN, Yalcin E, Dogru D, Yordam N. Neonatal hyperparathyroidism due to maternal hypoparathyroidism and vitamin D deficiency: a cause of multiple os fractures. Clin Pediatr. (2005) 44:267–ix. doi: 10.1177/000992280504400312
PubMed Abstract | CrossRef Full Text | Google Scholar
60. Bjørnerem Å, Bui Thousand, Wang X, Ghasem-Zadeh A, Hopper JL, Zebaze R, et al. Genetic and environmental variances of bone microarchitecture and os remodeling markers: a twin study. J Os Miner Res. (2015) 30:519–27. doi: 10.1002/jbmr.2365
PubMed Abstract | CrossRef Full Text | Google Scholar
61. Karasik D, Demissie S, Zhou Y, Lu D, Broe KE, Bouxsein ML, et al. Heritability and genetic correlations for bone microarchitecture: the framingham report families. J Bone Miner Res. (2017) 32:106–14. doi: 10.1002/jbmr.2915
PubMed Abstract | CrossRef Full Text | Google Scholar
62. Morris JA, Kemp JP, Youlten SE, Laurent 50, Logan JG, Chai RC, et al. An atlas of genetic influences on osteoporosis in humans and mice. Nat Genet. (2019) 51:258–66. doi: 10.1038/s41588-018-0302-10
CrossRef Full Text | Google Scholar
63. Collet C, Ostertag A, Ricquebourg K, Delecourt G, Tueur G, Isidor B, et al. Primary osteoporosis in young adults: Genetic basis and identification of novel variants in causal genes. JBMR Plus. (2017) ii:12–21. doi: ten.1002/jbm4.10020
PubMed Abstract | CrossRef Full Text | Google Scholar
65. Gregson CL, Wheeler L, Hardcastle SA, Appleton LH, Addison KA, Brugmans M, et al. Mutations in known monogenic high os mass loci just explain a small proportion of high os mass cases. J Bone Miner Res. (2016) 31:640–9. doi: 10.1002/jbmr.2706
PubMed Abstract | CrossRef Full Text | Google Scholar
66. Gregson CL, Newell F, Leo PJ, Clark GR, Paternoster Fifty, Marshall M, et al. Genome-broad clan report of farthermost loftier bone mass: contribution of common genetic variation to extreme BMD phenotypes and potential novel BMD-associated genes. Os. (2018) 114:62–71. doi: 10.1016/j.bone.2018.06.001
PubMed Abstruse | CrossRef Full Text | Google Scholar
67. Whyte MP, McAlister WH, Zhang F, Bijanki VN, Nenninger A, Gottesman GS, et al. New explanation for autosomal ascendant high bone mass: mutation of low-density lipoprotein receptor-related poly peptide 6. Bone. (2019) 127:228–43. doi: ten.1016/j.bone.2019.05.003
PubMed Abstract | CrossRef Full Text | Google Scholar
68. Gregson CL, Bergen DJM, Leo P, Sessions RB, Wheeler 50, Hartley A, et al. A rare mutation in SMAD9 associated with loftier bone mass identifies the SMAD-dependent BMP signaling pathway every bit a potential anabolic target for osteoporosis. J Bone Miner Res. (2019). doi: x.1002/jbmr.3875. [Epub alee of print].
PubMed Abstract | CrossRef Full Text | Google Scholar
70. Hmeljak J, Justice MJ. From gene to treatment: supporting rare illness translational research through model systems. Dis Model Mech. (2019) 12:dmm039271. doi: x.1242/dmm.039271
PubMed Abstruse | CrossRef Full Text | Google Scholar
72. Wangler MF, Yamamoto S, Chao HT, Posey JE, Westerfield Thou, Postlethwait J, et al. Model organisms facilitate rare disease diagnosis and therapeutic research. Genetics. (2017) 207:9–27. doi: x.1534/genetics.117.203067
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Kimmel DB. Fauna models in os research. In: Smith SY, Varela A, Samadfam R, editors. Bone Toxicology. Cham: Springer Nature (2017). p. 129–171. doi: 10.1007/978-3-319-56192-9_4
CrossRef Full Text | Google Scholar
74. Maynard RD, Ackert-Bicknell CL. Mouse models and online resources for functional assay of osteoporosis genome-wide association studies. Front Endocrinol. (2019) 10:277. doi: 10.3389/fendo.2019.00277
PubMed Abstract | CrossRef Full Text | Google Scholar
77. Bouxsein ML, Myers KS, Shultz KL, Donahue LR, Rosen CJ, Beamer WG. Ovariectomy-induced bone loss varies among inbred strains of mice. J Bone Miner Res. (2005) 20:1085–92. doi: 10.1359/JBMR.050307
PubMed Abstract | CrossRef Full Text | Google Scholar
78. Li Ten, Mohan S, Gu W, Wergedal J, Baylink DJ. Quantitative assessment of forearm muscle size, forelimb grip force, forearm bone mineral density, and forearm bone size in determining humerus breaking strength in 10 inbred strains of mice. Calcif Tissue Int. (2001) 68:365–nine. doi: 10.1007/s00223-001-0004-7
PubMed Abstract | CrossRef Total Text | Google Scholar
79. Lodberg A, Vegger JB, Jensen MV, Larsen CM, Thomsen JS, Brüel A. Immobilization induced osteopenia is strain specific in mice. Bone Rep. (2015) 2:59–67. doi: 10.1016/j.bonr.2015.04.001
PubMed Abstract | CrossRef Full Text | Google Scholar
lxxx. Lovell DP, Johnson FM, Willis DB. Quantitative genetic variation in the skeleton of the mouse: Ii. Clarification of variation inside and between inbred strains. Am J Anat. (1986) 176:287–303. doi: 10.1002/aja.1001760304
PubMed Abstract | CrossRef Total Text | Google Scholar
81. Sabsovich I, Clark JD, Liao Chiliad, Peltz M, Lindsey DP, Jacobs CR, et al. Bone microstructure and its associated genetic variability in 12 inbred mouse strains: microCT written report and in silico genome scan. Bone. (2008) 42:439–51. doi: 10.1016/j.os.2007.09.041
PubMed Abstract | CrossRef Total Text | Google Scholar
82. Fairfield H, Srivastava A, Ananda K, Liu R, Kircher 1000, Lakshminarayana A, et al. Exome sequencing reveals pathogenic mutations in 91 strains of mice with Mendelian disorders. Genome Res. (2015) 25:948–57. doi: 10.1101/gr.186882.114
PubMed Abstract | CrossRef Full Text | Google Scholar
83. Palmer K, Fairfield H, Borgeia S, Drape Thousand, Hassan MG, Dionne L, Yong Karst S, et al. Discovery and characterization of spontaneous mouse models of craniofacial dysmorphology. Dev Biol. (2016) 415:216–27. doi: ten.1016/j.ydbio.2015.07.023
PubMed Abstruse | CrossRef Full Text | Google Scholar
84. Brommage R, Powell DR, Vogel P. Predicting human illness mutations and identifying drug targets from mouse gene knockout phenotyping campaigns. Dis Model Mech. (2019) 12:5. doi: 10.1242/dmm.038224
PubMed Abstract | CrossRef Full Text | Google Scholar
85. Cacheiro P, Haendel MA, Smedley D, International Mouse Phenotyping Consortium and the Monarch Initiative. New models for human disease from the international mouse phenotyping consortium. Mamm Genome. (2019) xxx:143–l. doi: 10.1007/s00335-019-09804-five
PubMed Abstract | CrossRef Full Text | Google Scholar
86. Brommage R, Liu J, Vogel P, Mseeh F, Thompson AY, Potter DG, et al. NOTUM inhibition increases endocortical os formation and bone force. Bone Res. (2019) 7:2. doi: 10.1038/s41413-018-0038-iii
PubMed Abstract | CrossRef Full Text | Google Scholar
87. Brommage R, Liu J, Hansen GM, Kirkpatrick LL, Potter DG, Sands AT, et al. High-throughput screening of mouse gene knockouts identifies established and novel skeletal phenotypes. Bone Res. (2014) two:14034. doi: 10.1038/boneres.2014.34
PubMed Abstruse | CrossRef Full Text | Google Scholar
88. Iwaniec UT, Wronski TJ, Liu J, Rivera MF, Arzaga RR, Hansen G, et al. PTH stimulates bone formation in mice deficient in Lrp5. J Bone Miner Res. (2007) 22:394–402. doi: x.1359/jbmr.061118
PubMed Abstract | CrossRef Full Text | Google Scholar
89. Sawakami K, Robling AG, Ai 1000, Pitner ND, Liu D, Warden SJ, et al. The Wnt co-receptor LRP5 is essential for skeletal mechanotransduction but not for the anabolic bone response to parathyroid hormone treatment. J Biol Chem. (2006) 281:23698–711. doi: 10.1074/jbc.M601000200
CrossRef Full Text | Google Scholar
91. Roschger A, Roschger P, Keplingter P, Klaushofer M, Abdullah S, Kneissel M, et al. Event of sclerostin antibody treatment in a mouse model of severe osteogenesis imperfecta. Bone. (2014) 66:182–eight. doi: 10.1016/j.os.2014.06.015
PubMed Abstract | CrossRef Full Text | Google Scholar
92. Sinder BP, Eddy MM, Ominsky MS, Caird MS, Marini JC, Kozloff KM. Sclerostin antibody improves skeletal parameters in a Brtl/+ mouse model of osteogenesis imperfecta. J Os Miner Res. (2013) 28:73–fourscore. doi: 10.1002/jbmr.1717
PubMed Abstruse | CrossRef Full Text | Google Scholar
93. Jacobsen CM, Hairdresser LA, Ayturk UM, Roberts HJ, Deal LE, Schwartz MA, et al. Targeting the LRP5 pathway improves bone properties in a mouse model of osteogenesis imperfecta. J Bone Miner Res. (2014) 29:2297–306. doi: 10.1002/jbmr.2198
PubMed Abstract | CrossRef Full Text | Google Scholar
94. Petty DG, Peacock L, Mikulec Thou, Kneissel G, Kramer I, Cheng TL, et al. Combination sclerostin antibiotic and zoledronic acid treatment outperforms either treatment alone in a mouse model of osteogenesis imperfecta. Bone. (2017) 101:96–103. doi: 10.1016/j.bone.2017.04.016
PubMed Abstract | CrossRef Total Text | Google Scholar
95. Grafe I, Alexander Due south, Yang T, Lietman C, Homan EP, Munivez E, et al. Sclerostin antibiotic handling improves the bone phenotype of Crtap(-/-) mice, a model of recessive osteogenesis imperfecta. J Os Miner Res. (2016) 31:1030–40. doi: 10.1002/jbmr.2776
PubMed Abstract | CrossRef Full Text | Google Scholar
96. Ren Y, Han Ten, Jing Y, Yuan B, Ke H, Liu K, et al. Sclerostin antibody (Scl-Ab) improves osteomalacia phenotype in dentin matrix protein i (Dmp1) knockout mice with little impact on serum levels of phosphorus and FGF23. Matrix Biol. (2016) 52–4:151–61. doi: ten.1016/j.matbio.2015.12.009
CrossRef Full Text | Google Scholar
97. Kedlaya R, Veera South, Horan DJ, Moss RE, Ayturk UM, Jacobsen CM, et al. Sclerostin inhibition reverses skeletal fragility in an Lrp5-deficient mouse model of OPPG syndrome. Sci Transl Med. (2013) 5:211ra158. doi: 10.1126/scitranslmed.3006627
PubMed Abstract | CrossRef Full Text | Google Scholar
98. Choi JY, Lai JK, Xiong ZM, Ren Grand, Moorer MC, Stains JP, et al. Diminished canonical β-catenin signaling during osteoblast differentiation contributes to osteopenia in progeria. J Bone Miner Res. (2018) 33:2059–70. doi: x.1002/jbmr.3549
PubMed Abstract | CrossRef Total Text | Google Scholar
99. Bai Southward, Li D, Xu L, Duan H, Yuan J, Wei M. Recombinant mouse periostin ameliorates coronal sutures fusion in Twist1+/- mice. J Transl Med. (2018) 16:103. doi: 10.1186/s12967-018-1454-ii
PubMed Abstract | CrossRef Full Text | Google Scholar
100. He Y, Yuan C, Chen L, Liu Y, Zhou H, Xu Due north, et al. While information technology is not deliberate, much of today's biomedical research contains logical and technical flaws, showing a need for corrective action. Int J Med Sci. (2018) 15:309–22. doi: 10.7150/ijms.23215
CrossRef Total Text | Google Scholar
101. West DB, Engelhard EK, Adkisson M, Nava AJ, Kirov JV, Cipollone A, et al. Transcriptome analysis of targeted mouse mutations reveals the topography of local changes in gene expression. PLoS Genet. (2016) 12:e1005691. doi: 10.1371/journal.pgen.1005691
PubMed Abstract | CrossRef Total Text | Google Scholar
102. Maguire Due south, Estabel J, Ingham N, Pearson Due south, Ryder E, Carragher DM, et al. Targeting of Slc25a21 is associated with orofacial defects and otitis media due to disrupted expression of a neighbouring gene. PLoS ONE. (2014) 9:e91807. doi: 10.1371/journal.pone.0091807
PubMed Abstract | CrossRef Full Text | Google Scholar
103. Jokela H, Hakkarainen J, Kätkänaho 50, Pakarinen P, Ruohonen ST, Tena-Sempere M, et al. Deleting the mouse Hsd17b1 gene results in a hypomorphic Naglu allele and a phenotype mimicking a lysosomal storage disease. Sci Rep. (2017) 7:16406. doi: ten.1038/s41598-017-16618-v
PubMed Abstruse | CrossRef Full Text | Google Scholar
106. Heffner CS, Herbert Pratt C, Babiuk RP, Sharma Y, Rockwood SF, Donahue LR, et al. Supporting conditional mouse mutagenesis with a comprehensive Cre characterization resource. Nat Commun. (2012) three:1218. doi: x.1038/ncomms2186
PubMed Abstruse | CrossRef Full Text | Google Scholar
109. Bustin S, Nolan T. Talking the talk, but not walking the walk: RT-qPCR equally a image for the lack of reproducibility in molecular research. Eur J Clin Invest. (2017) 47:756–74. doi: 10.1111/eci.12801
CrossRef Full Text | Google Scholar
110. Abuna RPF, Oliveira FS, Ramos JIR, Lopes HB, Freitas GP, Souza ATP, et al. Option of reference genes for quantitative existent-time polymerase chain reaction studies in rat osteoblasts. J Jail cell Physiol. (2018) 234:749–56. doi: 10.1002/jcp.26886
PubMed Abstract | CrossRef Total Text | Google Scholar
111. Yang X, Hatfield JT, Hinze SJ, Mu 10, Anderson PJ, Powell BC. Bone to pick: the importance of evaluating reference genes for RT-qPCR quantification of gene expression in craniosynostosis and bone-related tissues and cells. BMC Res Notes. (2012) 5:222. doi: 10.1186/1756-0500-v-222
PubMed Abstract | CrossRef Full Text | Google Scholar
113. Saper CB. Editorial: an open alphabetic character to our readers on the use of antibodies. J Comp Neurol. (2005) 493:477–8. doi: 10.1002/cne.20839
CrossRef Total Text | Google Scholar
117. Hwang Prisoner of war, Horton JA. Variable osteogenic performance of MC3T3-E1 subclones impacts their utility as models of osteoblast biology. Sci Rep. (2019) 9:8299. doi: 10.1038/s41598-019-44575-8
PubMed Abstract | CrossRef Full Text | Google Scholar
118. Williams DK, Pinzón C, Huggins S, Pryor JH, Falck A, Herman F, et al. Genetic engineering a large animal model of human hypophosphatasia in sheep. Sci Rep. (2018) viii:16945. doi: x.1038/s41598-018-35079-y
PubMed Abstract | CrossRef Full Text | Google Scholar
119. Kyöstilä K, Syrjä P, Lappalainen AK, Arumilli M, Hundi S, Karkamo Five, et al. A homozygous missense variant in the alkaline phosphatase factor ALPL is associated with a severe form of canine hypophosphatasia. Sci Rep. (2019) ix:973. doi: 10.1038/s41598-018-37801-2
PubMed Abstruse | CrossRef Full Text | Google Scholar
120. Packer RA, Logan MA, Guo LT, Apte SS, Bader H, O'Brien DP, et al. Clinical phenotype of Musladin-Lueke syndrome in 2 beagles. J Vet Intern Med. (2017) 31:532–8. doi: x.1111/jvim.14654
PubMed Abstruse | CrossRef Full Text | Google Scholar
121. Quist EM, Doan R, Puddle RR, Porter BF, Bannasch DL, Dindot SV. Identification of a candidate mutation in the COL1A2 gene of a grub with osteogenesis imperfecta. J Hered. (2018) 109:308–14. doi: ten.1093/jhered/esx074
PubMed Abstruse | CrossRef Total Text | Google Scholar
122. Mansour TA, Lucot K, Konopelski SE, Dickinson PJ, Sturges BK, Vernau KL, et al. Whole genome variant association across 100 dogs identifies a frame shift mutation in DISHEVELLED 2 which contributes to Robinow-like syndrome in Bulldogs and related spiral tail dog breeds. PLoS Genet. (2018) 14:e1007850. doi: 10.1371/journal.pgen.1007850
PubMed Abstract | CrossRef Full Text | Google Scholar
123. Willet CE, Makara Thousand, Reppas Grand, Tsoukalas G, Malik R, Haase B, et al. Canine disorder mirrors human disease: exonic deletion in HES7 causes autosomal recessive spondylocostal dysostosis in miniature Schnauzer dogs. PLoS ONE. (2015) x:e0117055. doi: 10.1371/journal.pone.0117055
PubMed Abstruse | CrossRef Full Text | Google Scholar
124. Lindert U, Weis MA, Rai J, Seeliger F, Hausser I, Leeb T, et al. Molecular consequences of the SERPINH1/HSP47 mutation in the dachshund natural model of osteogenesis imperfecta. J Biol Chem. (2015) 290:17679–89. doi: 10.1074/jbc.M115.661025
PubMed Abstract | CrossRef Full Text | Google Scholar
125. Yin Y, Bangs F, Paton IR, Prescott A, James J, Davey MG, et al. The Talpid3 gene (KIAA0586) encodes a centrosomal poly peptide that is essential for master cilia formation. Development. (2009) 136:655–64. doi: 10.1242/dev.028464
PubMed Abstract | CrossRef Full Text | Google Scholar
126. Maas SA, Suzuki T, Fallon JF. Identification of spontaneous mutations within the long-range limb-specific Sonic hedgehog enhancer (ZRS) that modify Sonic hedgehog expression in the chicken limb mutants oligozeugodactyly and silkie breed. Dev Dyn. (2011) 240:1212–22. doi: 10.1002/dvdy.22634
PubMed Abstract | CrossRef Full Text | Google Scholar
128. Lleras-Forero L, Winkler C, Schulte-Merker S. Zebrafish and medaka equally models for biomedical enquiry of os diseases. Dev Biol. (2019). doi: ten.1016/j.ydbio.2019.07.009. [Epub ahead of impress].
PubMed Abstract | CrossRef Full Text | Google Scholar
129. Howe Thou, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato Yard, et al. The zebrafish reference genome sequence and its relationship to the human being genome. Nature. (2013) 496:498–503. doi: x.1038/nature12111
PubMed Abstract | CrossRef Total Text | Google Scholar
130. Briggs MD, Bell PA, Wright MJ, Pirog KA. New therapeutic targets in rare genetic skeletal diseases. Exp Opin Orphan Drugs. (2015) 3:1137–1154. doi: ten.1517/21678707.2015.1083853
PubMed Abstruse | CrossRef Full Text | Google Scholar
131. Semler O, Rehberg M, Mehdiani N, Jackels M, Hoyer-Kuhn H. Electric current and emerging therapeutic options for the management of rare skeletal diseases. Paediatr Drugs. (2019) 21:95–106. doi: ten.1007/s40272-019-00330-0
PubMed Abstract | CrossRef Full Text | Google Scholar
132. Canalis E, Sanjay A, Yu J, Zanotti South. An antibiotic to Notch2 reverses the osteopenic phenotype of Hajdu-Cheney mutant male person mice. Endocrinology. (2017) 158:730–42. doi: ten.1210/en.2016-1787
PubMed Abstruse | CrossRef Full Text | Google Scholar
133. Howles SA, Hannan FM, Gorvin CM, Piret SE, Paudyal A, Stewart Grand, et al. Cinacalcet corrects hypercalcemia in mice with an inactivating Gα11 mutation. JCI Insight. (2017) 2:xx. doi: 10.1172/jci.insight.96540
PubMed Abstract | CrossRef Full Text | Google Scholar
134. Khan T, Sinkevicius KW, Vong S, Avakian A, Leavitt MC, Malanson H, et al. ENPP1 enzyme replacement therapy improves blood pressure level and cardiovascular office in a mouse model of generalized arterial calcification of infancy. Dis Model Mech. (2018) 11:dmm035691. doi: x.1242/dmm.035691
PubMed Abstract | CrossRef Total Text | Google Scholar
136. Wu Northward, Liu B, Du H, Zhao South, Li Y, Cheng 10, et al. The progress of CRISPR/Cas9-mediated gene editing in generating mouse/zebrafish models of human skeletal diseases. Comput Struct Biotechnol J. (2019) 17:954–62. doi: 10.1016/j.csbj.2019.06.006
PubMed Abstruse | CrossRef Total Text | Google Scholar
138. Gonorazky HD, Naumenko Due south, Ramani AK, Nelakuditi V, Mashouri P, Wang P, et al. Expanding the boundaries of RNA sequencing as a diagnostic tool for rare Mendelian affliction. Am J Hum Genet. (2019) 104:466–83. doi: 10.1016/j.ajhg.2019.01.012
CrossRef Total Text | Google Scholar
139. Khayal LA, Grünhagen J, Provazník I, Mundlos S, Kornak U, Robinson PN, et al. Transcriptional profiling of murine osteoblast differentiation based on RNA-seq expression analyses. Bone. (2018) 113:29–xl. doi: ten.1016/j.bone.2018.04.006
PubMed Abstract | CrossRef Total Text | Google Scholar
140. Sebastian A, Hum NR, Morfin C, Murugesh DK, Loots GG. Global factor expression analysis identifies Mef2c as a potential player in Wnt16-mediated transcriptional regulation. Gene. (2018) 675:312–21. doi: 10.1016/j.cistron.2018.06.079
PubMed Abstract | CrossRef Full Text | Google Scholar
Source: https://www.frontiersin.org/articles/10.3389/fendo.2019.00934/full
Posted by: wilsonthisity93.blogspot.com
0 Response to "What Effects Do You Think Skeletal Disorders Have On Animals?"
Post a Comment